Transition metal–catalyzed enantioselective cycloaddition of cyclopropanes with π-unsaturated compounds represents one of the most important strategies for constructing chiral five-membered ring structures. However, owing to the high C–C bond energy of cyclopropanes, their activation is intrinsically challenging. Consequently, existing methods are largely confined to cyclopropanes bearing high ring strain or strongly electron-withdrawing substituents (Figure 1a, Type I), and are difficult to apply to weakly activated cyclopropyl ketones that are inexpensive, readily available, and synthetically versatile (Figure 1a, Type II). The central challenge in achieving highly enantioselective cycloaddition of cyclopropyl ketones lies in the strong electrophilicity of the ketone carbonyl group. Under the elevated temperatures or Lewis acid conditions typically required for C–C bond cleavage, the chiral α-tertiary carbon center in the product is difficult to maintain and is prone to racemization (Figure 1b). As a result, although racemic cycloaddition reactions of cyclopropyl ketones have been successfully developed since 2006, highly enantioselective variants have remained elusive. To address this issue, several substrate-modification strategies have been developed. For example, replacing the ketone with a less electrophilic carboxamide group enables highly enantioselective cycloaddition with alkynes at 130 °C (Figure 1b, left). Alternatively, the use of multisubstituted cyclopropanes to stabilize radical intermediates has allowed the development of radical-type cycloaddition reactions under mild conditions (Figure 1b, right). Although these approaches can achieve high enantioselectivity, the specific structural requirements of the substrates significantly limit product diversity and thereby restrict the scope of application. Therefore, achieving efficient asymmetric cycloaddition of simple cyclopropyl ketones through catalyst control remains a key and long-standing challenge in this field.
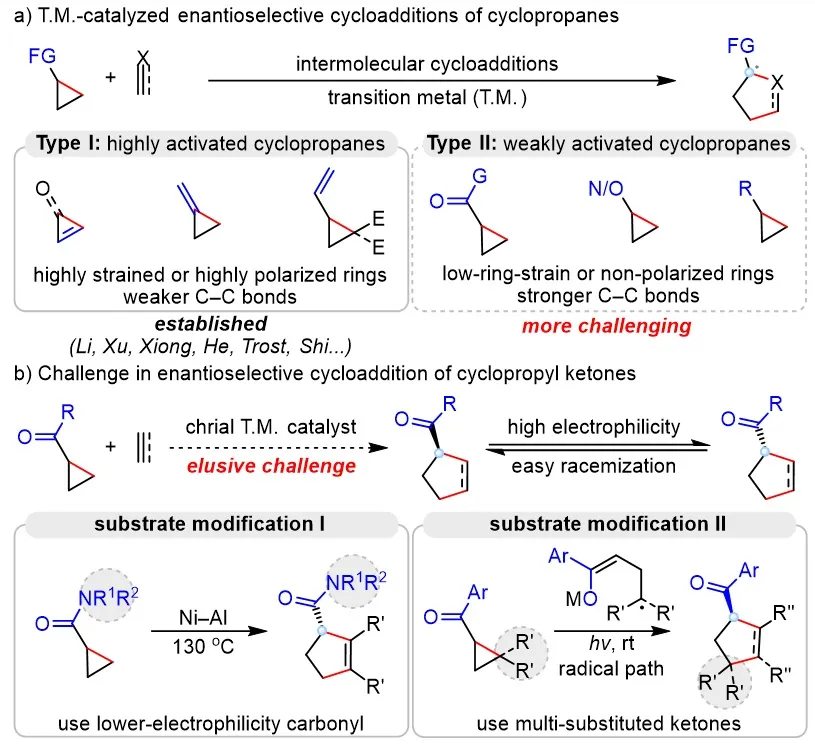
Figure 1. Background
Recently, the Mengchun Ye / Weiwei Xu team at Nankai University proposed an innovative catalytic strategy. By designing a new class of chiral diamine–phosphine oxide (PO) ligands, they achieved a marked enhancement in the reactivity of this cycloaddition reaction, enabling it to proceed efficiently even at room temperature, thereby effectively mitigating product racemization. In addition, the introduction of weakly coordinating tetrahydrofuran (THF) as the solvent further attenuates the Lewis acidity of the aluminum center, suppressing product racemization. As a result, under mild conditions (room temperature), a highly enantioselective [3 + 2] cycloaddition of cyclopropyl ketones with alkynes was successfully realized. The reaction not only exhibits broad substrate scope but also delivers yields of up to 99% and enantioselectivities of up to 99% ee (Figure 2). Mechanistic studies indicate that the high reactivity and enantioselectivity mainly arise from three key factors: the high activity of the newly developed PO ligand, the moderately attenuated Lewis acidity achieved through synergistic PO/THF modulation, and the mild reaction temperature. The concerted action of these factors effectively suppresses enolization-induced racemization pathways while avoiding side reactions caused by the high electrophilicity of the ketone carbonyl group. Relevant achievements were published in J. Am. Chem. Soc. 2025, DOI: 10.1021/jacs.5c18087.
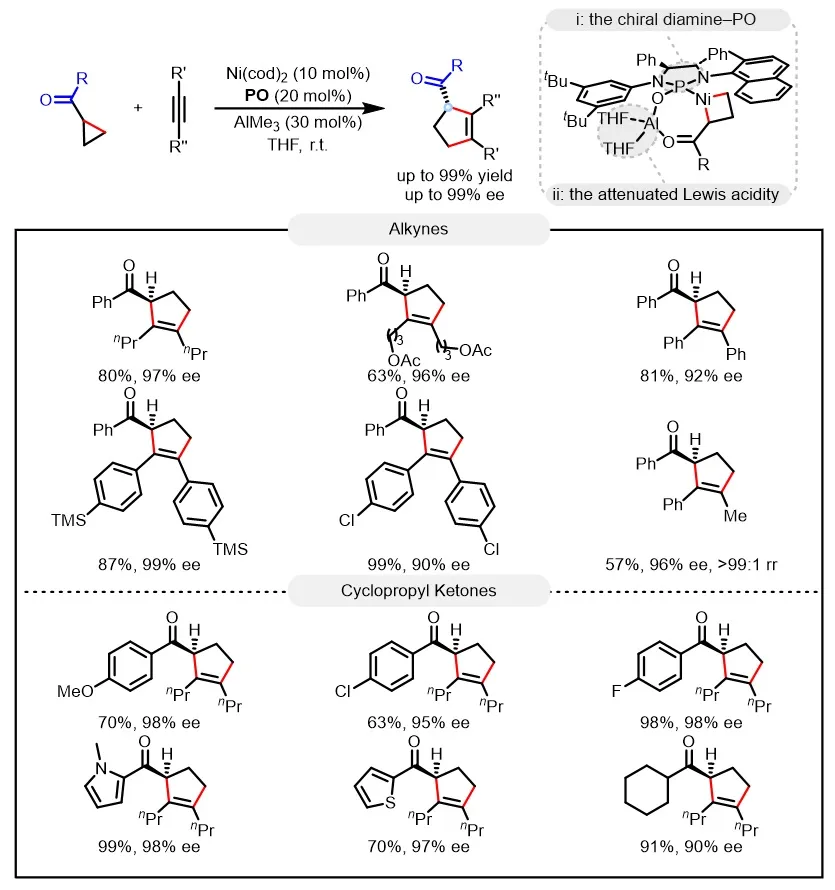
Figure 2. Ni/Al bimetal-catalyzed enantioselective cycloaddition of cyclopropyl ketones


