A sustainable carbon cycle economy based on solar energy is crucial for the development of future society. Photocatalytic CO2 reduction (PCO2R) to syngas has been considered a promising proposal in the direct utilization of solar energy. Syngas (the mixture of H2 and CO) is a key feedstock for accessing numerous value-added chemicals/fuels in the standard industrial processes (e.g., Fischer–Tropsch synthesis and hydroformylation). Current photocatalytic CO2-to-syngas systems often require sacrificial agents (e.g., triethanolamine) to scavenge photogenerated holes for enhanced efficiency, compromising sustainability and economic viability. Although heterogeneous catalysts enable coupled CO2 reduction with organic oxidation, their poorly understood reactive sites hinder reaction diversification. In contrast, homogeneous molecular systems offer well-defined structures for the rational design of coupled reactions. However, catalytic systems or catalysts integrating CO2 reduction and organic oxidation reactions still remain scarce, due to severe back electron transfer in homogeneous photocatalysis. Catalytic dehydrogenation of saturated organic compounds, e.g., N-heterocyclic compounds, coupled with CO2 reduction to produce unsaturated molecules and syngas, is a fundamentally important chemical process with a wide application in organic synthesis and for a potential future “carbon-recycling economy society”. However, this process is usually very difficult because the desaturation of organic compounds is generally unfavorable in terms of the enthalpy factor, and also CO2 is a thermodynamically very stable and kinetically inert molecule. Although in the classical proton-coupled electron transfer (PCET) process, the feasibility of the coupled reaction can be assessed by redox potential and pKa parameters. In previous reports, N-heterocyclic compound dehydrogenation coupled with CO2 reduction reactions are dependent on heterogeneous rare metal materials such as InP-In2O3 heterojunctions, or Ru supported-MOF-235. Since the formation of more redox-active species increases the chance of potential back electron transfer, it in turn leads to some issues such as low reaction efficiency and difficulty in obtaining stoichiometric dehydrogenation and reduction products. In contrast, hydrogen atom transfer (HAT) can simultaneously render the transfer of electron and proton by only one-step reaction, which avoids the formation of active species and thus effectively improves the reaction efficiency and product selectivity.
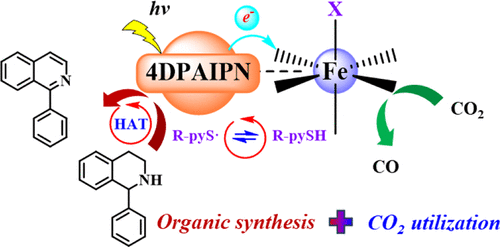
Recently, Liang-Nian He’s group have developed a pyridinethiol iron-catalyzed system to achieve N-heterocyclic dehydrogenation coupled with CO2 photoreduction, efficiently delivering completely dehydrogenated isoquinoline products along with a stoichiometric ratio of syngas via synergistic cyanoaromatic photoredox catalysis, earth-abundant metal iron-catalysis, and pyridinethiol organo-catalytic units. The mechanistic studies revealed that the reductive quenching of the photosensitizer 4DPAIPN* by pyridinethiol serves as the initiating step, and the subsequent HAT process mediated by the pyridinethiol radical functions as the key step depending on the S–H bond dissociation enthalpies and the radical polarity to facilitate the coupled reaction. The establishment of this system will be helpful to further explore and develop economical, environmentally friendly, and sustainable CO2 photoreduction solutions. Relevant achievements were published in J. Am. Chem. Soc., 2025. 147. 31482-31487.


