Recently, Weiwei Zee's group at Nankai University developed a copper/palladium synergistic catalytic system to achieve enantioselective Csp2-Csp3 coupling reactions of dienyl fluorides with aldimine esters and obtained a series of highly enantioselective chiral α-alkenyl α-amino acid derivatives. Notably, this is the first case of asymmetric Csp2-Csp3 cross-coupling of an inert Csp2-F bond. Controlled experiments and DFT computational studies have shown that the reaction is activated by Pd-H migration insertion and subsequent allyl defluorination process, rather than the direct oxidative addition of Csp2-F bond to Pd(0). Article links. DOI:10.1038/s41467-022-30152-7
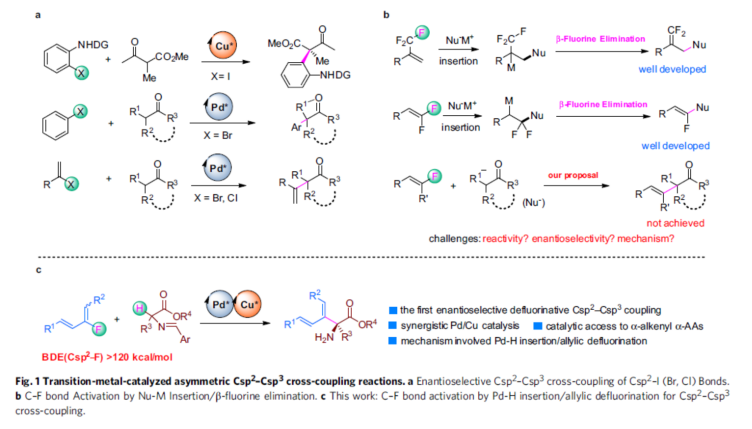
Transition-metal-catalyzed enantioselective cross-coupling reactions between Csp2–X compounds (X = halogen) and enolizable carbonyl compounds are commonly used transformations for asymmetric construction Csp2–Csp3 bonds. Many successful examples of this method have been reported, including pioneering work by the research groups of Ma, Hartwig, and Buchwald, who used chiral-ligand-bearing transition metals such as Cu and Pd to achieve enantiocontrol(Fig. 1a). These reactions initiated with oxidative addition of Csp2–X bond to the Pd(0) or Cu(I), followed by ligand exchange and reductive elimination to form the Csp2–Csp3 bond. Halogenated compounds with Csp2–I, Csp2–Br, and even Csp2–Cl bonds are suitable reaction substrates. However, because Csp2–F bonds have high energies (120–129 kcal/mol for olefinic C–F bonds), fluorinated compounds have rarely been used as coupling partners.
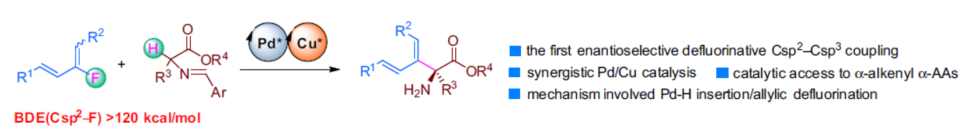
Recently, C–F activation has been an important research topic in synthetic organic chemistry. One of the most successful strategies in this area is using transition-metal-mediated or -catalyzed metal-Nu insertion/β–F elimination process. Trifluoro-, difluoroalkenes have been intensively investigated for this purpose during the past few years; however, monofluoroalkenes are rarely explored class of compounds for similar C–F activation reactions (Fig. 1b).In this work, we report a Cu/Pd cooperative system for enantioselective Csp2–Csp3 cross-coupling between dienyl fluorides and aldimine esters (Fig. 1c). Experimental and computational studies revealed that this reaction involved a unique Pd-H insertion/allylic defluorination process. This work not only represents the first example of enantioselective defluorinative Csp2–Csp3 coupling but also provides a highly efficient catalytic method to prepare chiral α-alkenyl α-amino acids (α-AAs), which are important synthetic targets because of their potential biochemical and pharmacological activities. Experimental and computational studies have shown that the reaction involves a unique process of Pd-H insertion/allyl defluorination.
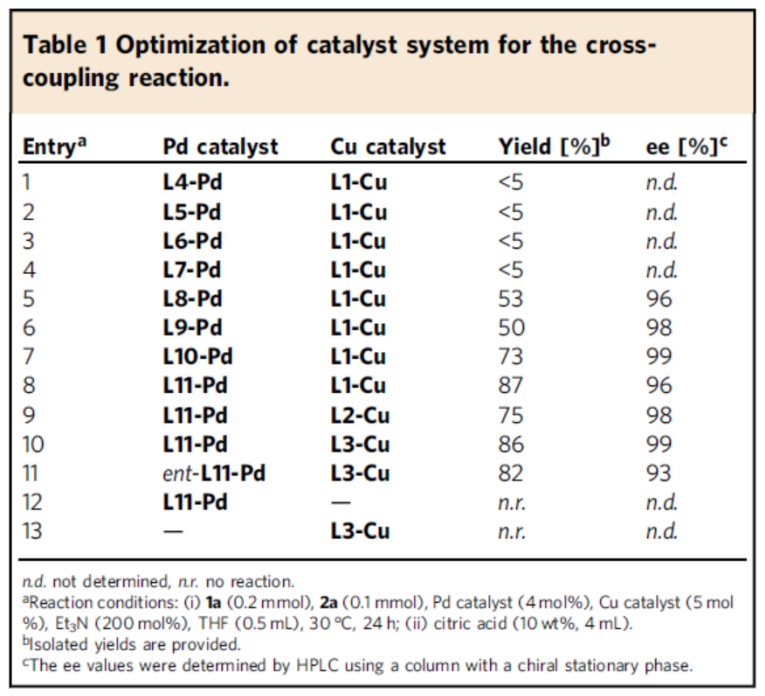
The studies begin with investigating the reaction of dienyl fluoride E-1a with aldimine ester 2a to generate α-alkenyl, α-methyl α-AA 3aa. Coordinative catalitic system L11-Pd/L3-Cu gave the best results (86% yield, 99% ee). At the same time, the enantioselectivity of the reaction is mainly controlled by the chiral Cu catalyst.
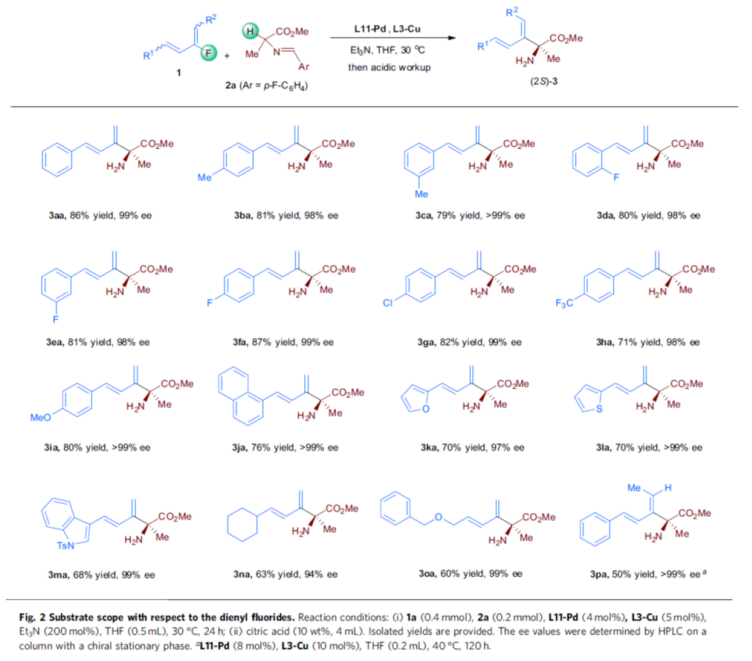
Having developed an effective dual-metal catalyst system, the authors investigated the substrate scope of the reaction, starting with dienyl fluorides 1 bearing various R1 and R2 substituents (Fig. 2). First, when R1 of substrate 1 is a series of phenyl and heteroaryl groups containing different electrical substitutions, they react smoothly with 2a, and the corresponding product 3aa-3ma is obtained, with a yield of 68-87%, ee is 98->99%. Secondly, when R1 in substrate 1 is a cyclohexyl group, the product 3na can be obtained at a yield of 63% and 94% ee. At the same time, the substrate with an allyl BnO ether unit can also be compatible with the system, and the product 3oa is obtained, with a yield of 60% and ee of 99%. In addition, 1,4-di-substituted dialenyl fluoride (R1 = Ph, R2 = Me), is also a suitable substrate, obtaining a product of 50% yield and > 99% ee 3pa.

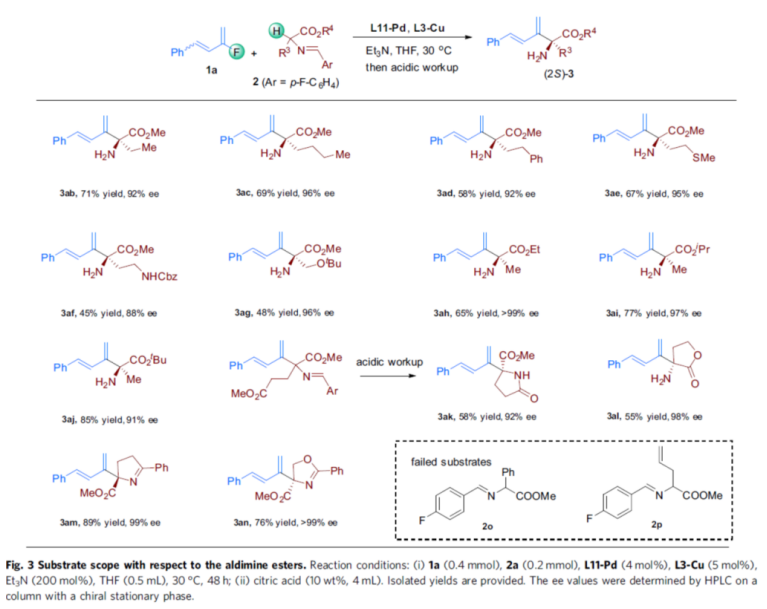
Immediately afterwards, the authors extended the range of aldimine ester substrates 2 (Fig. 3). First, when R4 in substrate 2 is methyl and R3 is ethyl, n-butyl, phenylethyl, and alkyl containing heteroatoms, all reacted smoothly with 1a to obtain the corresponding products 3ab-3ag in 45-71% yield and 88-96% ee. Secondly, when R3 in substrate 2 is methyl and R4 is ethyl, isopropyl and tert-butyl, it is also compatible with the system to obtain the corresponding products 3ah-3aj in 65-85% yield and 91->99% ee. Meanwhile, the aldimine ester substrate derived from glutamic acid, was also able to react smoothly, but the reaction underwent cyclization due to acidic post-treatment, resulting in the lactam product 3ak in 58% yield and 92% ee. α-Amino-γ-butyrolactone-derived imine substrate could also react with 1a to obtain the cyclized product 3al in 55% yield and 98% ee. In addition, cyclic ketimine esters and oxazolyl esters that are also suitable substrates, such as 3am and 3an. however, the phenylalanine-derived aldimine ester 2o (affected by spatial site blocking) failed to react successfully with the allyl-containing substrate 2p (possibly affected by partial isomerization of the terminal olefin).
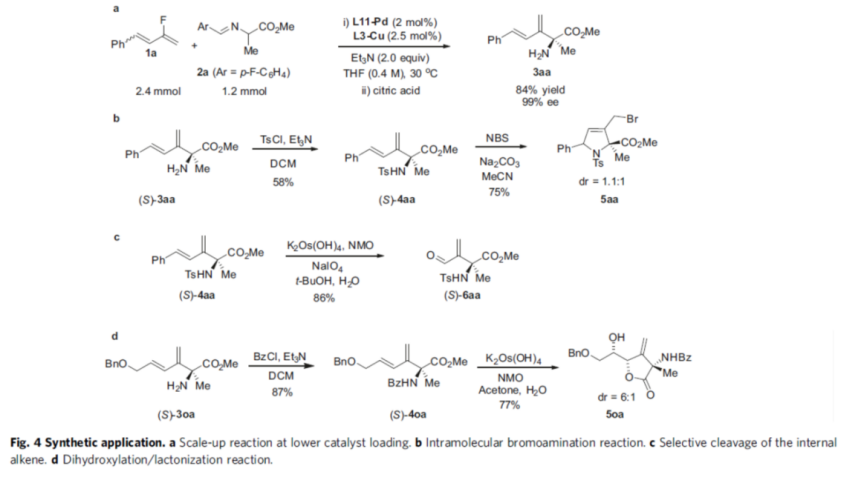
Also, the authors investigated the practicality of the reaction (Fig. 4). Firstly, the reaction size was scaled up to more than 1 mmol when the catalyst loading could be further reduced and the reaction results were not affected (Fig. 4a). Second, the amine in (S)-3aa was protected by Ts and then reacted in acetonitrile solvent of NBS/Na2CO3 to obtain the brominated amination product 5aa in 75% yield (Fig. 4b). The inner olefin unit in (S)-4aa can be selectively broken using K2Os(OH)4/NMO followed by NaIO4 treatment to obtain the aldehyde product (S)-6aa in 86% yield (Fig. 4c). The amino group in (S)-3oa was protected by Bz and then dihydroxylated/lactonized to obtain the lactone compound 5oa in good diastereoselectivity (Fig. 4d).
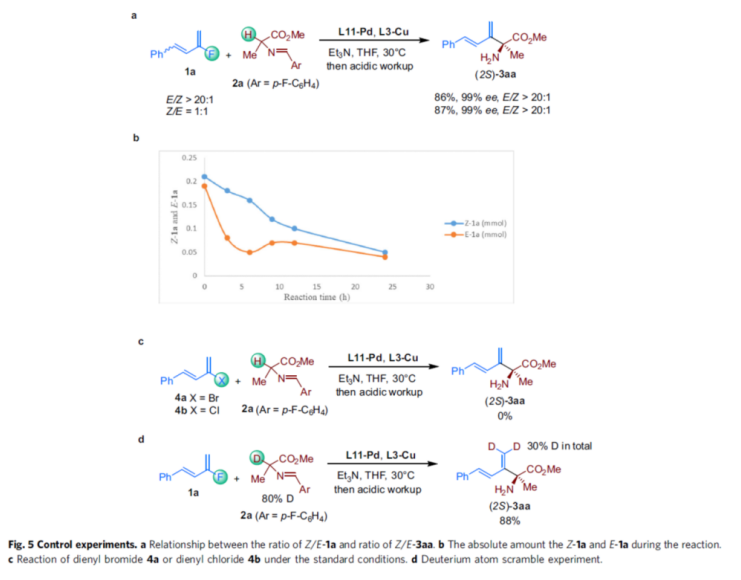
To further understand the mechanism of the reaction, the authors performed relevant controlled experiments (Fig. 5). First, the reaction of 1a with E/Z of >20:1 versus 1:1 with 2a, respectively, resulted in the product (2S)-3aa in similar yields with enantioselectivity (Fig. 5a). Meanwhile, when the consumption of Z-1a and E-1a in 1a (E/Z of 1:1.1) was monitored with the reaction time, it was found that the concentration of both Z-1a and E-1a decreased during the reaction, but the consumption of E-1a was faster, and the difference in consumption between the two was the largest after 5 h (Fig. 5b). The above results suggest that E-1a may react preferentially and that Z-1a gradually isomerizes to E-1a. Second, when dienyl bromide 4a or dienyl chloride 4b was used as the substrate, the desired coupling product 3aa failed to be obtained under the above conditions, thus excluding the direct oxidative addition pathway of the Csp2-F bond to Pd(0) (Fig. 5c). Furthermore, the reaction of 1a with deuterium-labeled 2a revealed a 30% deuterium doping on the terminal carbon atom of the double bond in the product, thus indicating that the reaction may involve a Pd-D insertion process (Fig. 5d).
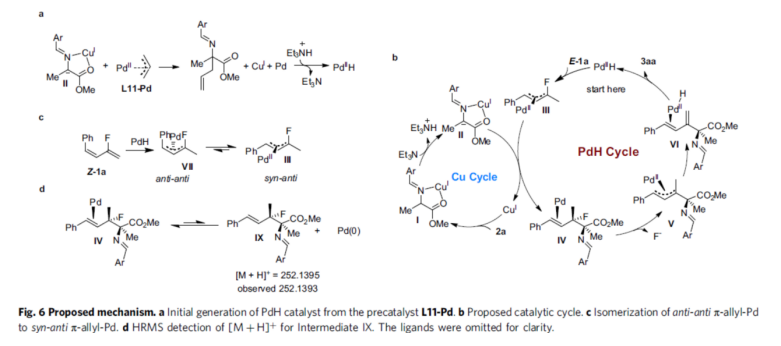
Based on the above study and the review of related literature, the authors proposed a rational catalytic cycling process (Fig. 6). First, in the Cu catalytic cycle, Cu acts as Lewis acid to activate the aldimine ester 2a to generate the yelid intermediate II, which acts as a nucleophilic reagent in the coupling reaction. II is allylicized with L11-Pd to generate L11-Pd(0), which can undergo oxidative addition with Et3NH+ to generate the Pd-H catalyst (Fig. 6a). In the Pd-H catalytic cycle (Fig. 6b), Pd-H migrates and inserts into the C=C bond of the fluoroethylene unit in E-1a to generate the fluorinated Pd-allyl intermediate III. III and II undergo allylic substitution to generate intermediate IV and regenerate the copper catalyst. IV undergoes a rapid allylic defluorination process to generate the allyl-Pd intermediate V, which undergoes β-H elimination to generate intermediate VI. Finally, VI was dissociated to give product 3aa and the Pd-H catalyst was regenerated. For Z-1a, anti-anti η3-Pd-allyl intermediate VII is generated after Pd-H migration insertion and is equilibrated to the thermodynamically more stable syn-anti η3-Pd-allyl intermediate III via η3-η1-η3 isomerization (Fig. 6c). Thus, Z-1a would undergo the same catalytic cycle to produce the same E-type product 3aa. In addition, the reaction was found in the HRMS spectrum of the reaction mixture to form intermediate IX, derived from the dissociation of Pd(0) from intermediate IV (Fig. 6d).
(from:Nat. Commun.)
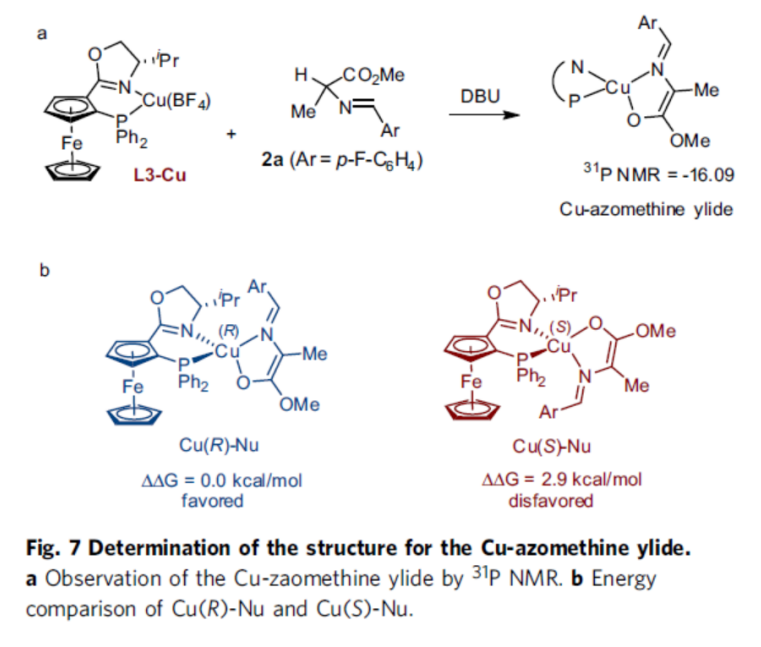
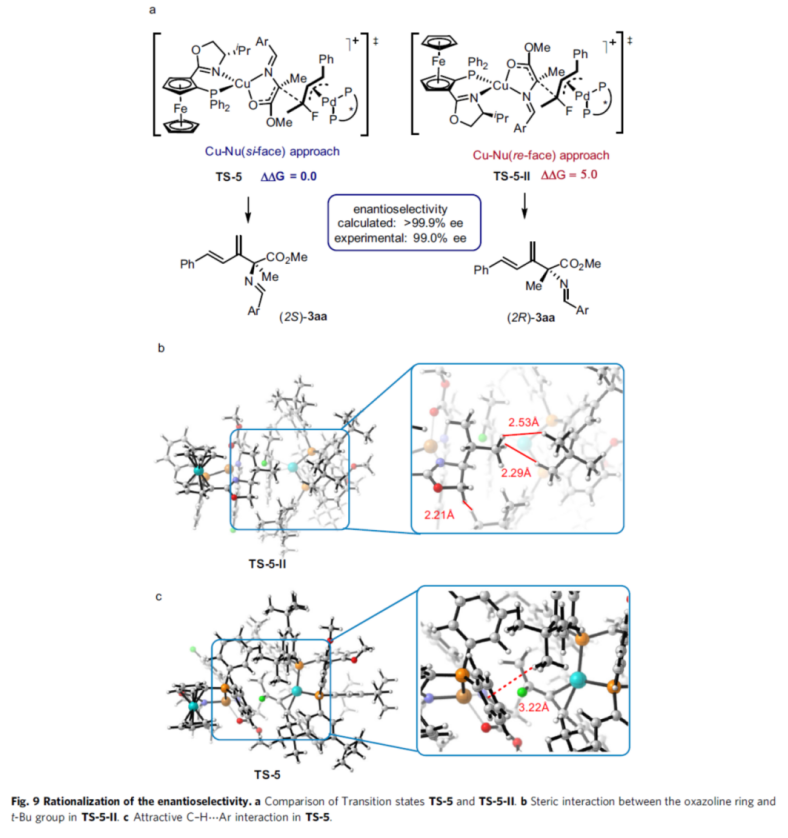
Finally, the authors further justified the above mechanism by the relevant DFT calculations (Fig. 7-9).
(from:Nat. Commun.)

(from:Nat. Commun.)
In conclusion: Weiwei Zi's group at Nankai University has developed a copper/palladium synergistic catalytic system to achieve the first enantioselective Csp2-Csp3 coupling reaction of dienyl fluorides with aldimine esters and to obtain a series of chiral α-alkenyl α-amino acid derivatives with a wide range of substrates, high functional group compatibility and excellent enantioselectivity. Control experiments and DFT calculation studies showed that the reactions were initiated by Pd-H migration insertion and generated C-C bonds via nucleophilic allylic substitution. This is followed by a Pd/Et3NH+ mediated allylic defluorination and β-H elimination process, leading to the generation of the coupling product and regeneration of the Pd-H catalyst.


