The construction of chiral quaternary carbons has received intensive attention in recent decades, because chiral quaternary carbons are prevalent structural units in numerous natural products, pharmaceuticals, and agrochemicals. Among existing approaches, enantioselective transition metal-catalyzed C–H alkylation represents one of the most atom- and step-economical routes to chiral quaternary carbons, owing to relatively easily-accessed starting materials and minimum loss of atoms during reactions (Scheme 1a). Though appealing, this process often suffers from some big challenges such as low reactivity of inert C–H bond activation, difficult insertion of sterically-hindered 1,1-disubstituted alkenes into the formed C–M–H intermediates, as well as tricky control of regioselectivity and enantioselectivity. To ease the process, the selection of C–H bonds and their activating method are critical. A big progress has been made for aryl C–H alkylations, in which relatively reactive heteroaryl C–H bonds or unreactive aryl C–H bonds are activated by Pd, Rh, Sc, and Ir, providing the corresponding aryl C–M intermediates that are stable and can be further inserted by 1,1-disubstitutedalkenes to give final quaternary carbons with high ee (Scheme 1b, left). Another important advance is achieved by Yu and Suginome, who use allylic C–H bonds or C–H bonds adjacent to heteroatoms to generate alkyl C–M intermediates, which are also stable enough to be trapped by 1,1-disubstituted alkenes, furnishing chiral quaternary carbons with high ee (Scheme 1b, right). Nevertheless, the activation of acyl C–H bonds to form chiral quaternary carbons bearing easily-transformable carbonyl groups still remains an elusive challenge, despite wide existence of such structural motifs in bioactive molecules. A possible reason for this challenge is that unstable C(O)–M intermediates may undergo decarbonylation to release CO species, which will not only inhibit metal activity, but also hinder chiral ligands from coordinating with metals, resulting in low selectivity. To address this issue, Chang and coworkers use a Ru catalyst to explore formyl C–H alkylation, achieving oxindoles bearing a sterically-hindered quaternary carbon as main products (Scheme 1c). Despite a big breakthrough in acyl C–H alkylation, the lack of proper chiral ligands renders the control of regioselectivity and enantioselectivity quite difficult.
Recently, Mengchun Ye’s group successfully constructed chiral quaternary carbons bearing a formyl group via a highly regioselective and highly enantioselective bimetal-catalyzed C–H alkylation of formamides with alkenes under mild conditions. A naphthylamine-derived phosphine oxide-ligated Ni and Al bimetallic catalyst was identified as the optimal catalyst. A wide range of di-, tri- and even tetra-substituted alkenes were compatible with the reaction, affording oxindoles bearing a quaternary carbon in up to 94%yield and up to 95% ee. Versatile transformations of amide group, oxindole and aryl ring demonstrates potential utility of the method. Relevant achievements were published in Angew. Chem. Int. Ed., 2024, DOI: 10.1002/anie.202413652.
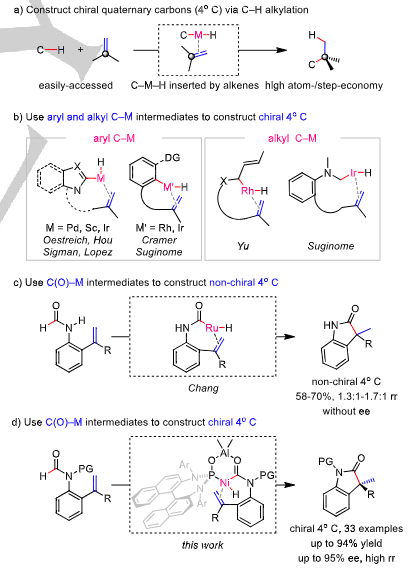
Scheme 1. Construct oxindoles bearing a chiral quaternary carbon via enantioselective transition metal-catalyzed C–H alkylation.


