Organoboron compounds have a broad range of applications, and the development of efficient methods for their synthesis is attracting intense interest. Although several powerful methods, such as Brown’s hydroboration of olefins, transition-metal-catalyzed hydroboration of olefins, Miyaura’s borylation reaction, and direct borylation of C–H bonds, have been established, new efficient reactions for C–B bond formation remain highly desirable to meet the requirements of the diverse syntheses being attempted today.
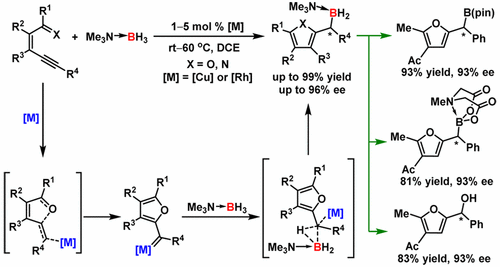
Prof. Zhou and Prof. Zhu report transition-metal-catalyzed B–H bond insertion reactions between borane adducts and alkynes to afford organoboron compounds in excellent yields under mild reaction conditions. This successful use of alkynes as carbene precursors in these reactions constitutes a new route to organoboron compounds. The starting materials are safe and readily available, and the reaction exhibits 100% atom-economy. Moreover, an asymmetric version catalyzed by chiral dirhodium complexes produced chiral boranes with excellent enantioselectivity (up to 96% ee). This is the first report of highly enantioselective heteroatom–hydrogen bond insertion reactions of metal carbenes generated in situ from alkynes. The chiral products of the reaction could be easily transformed to widely used borates and diaryl methanol compounds without loss of optical purity, which demonstrates its potential utility in organic synthesis. A kinetics study indicated that the Cu-catalyzed B–H bond insertion reaction is first order with respect to the catalyst and the alkyne and zero order with respect to the borane adduct, and no kinetic isotopic effect was observed in the reaction of the adduct. These results, along with density functional theory calculations, suggest that the formation of the Cu carbene is the rate-limiting step and that the B–H bond insertion is a fast, concerted process.
Link: http://pubs.acs.org/doi/abs/10.1021/jacs.6b13168


