Marine natural products are important sources for novel drug discoveries. They are divergent in both chemical structures and biological activities. In 2009, Sato and co-workers isolated a class of novel polyketides from marine-derived actinomyces, which they named indoxamycins A-F. This family of natural products shares a tricyclic caged [5.5.6] structure with a tri-substituted alkene and an α,β
-unsaturated acid serving as side chains. A prominent feature of these molecules is their dense array of 6 contiguous stereogenic centers containing 3 all-carbon quaternary centers, 2 of which are vicinal. The synthetic challenge posed by the complex structure and the anti-tumors activities matching the potency of the traditional chemotherapy drug mitomycin has made these molecules hot targets among synthetic chemists and medicinal chemists. In 2012, Carreira and co-workers first published a total synthesis of racemic indoxamycin B (
Angew. Chem. Int. Ed.,
2012
,
51
, 3474). In 2013 and 2014, the Ding group reported the asymmetric syntheses of (-)-indoxamycins A-F
(Angew. Chem. Int. Ed., 2013, 52, 13256; Chem. Eur. J., 2014, 20, 15053)。
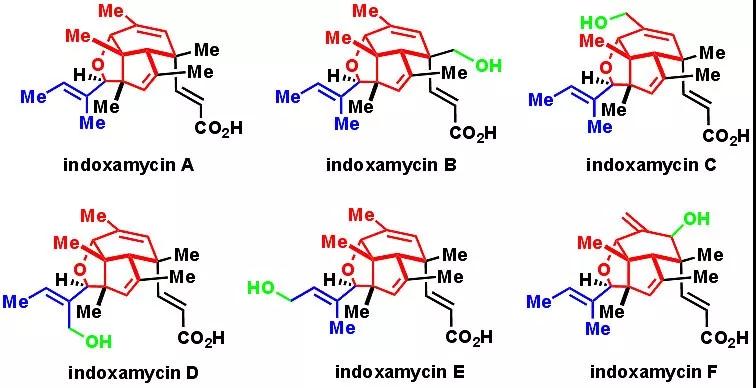
Recently, Liang et al demonstrated a novel concise route towards these natural products on Angew. Chem. Int. Ed. The first author of the article was Dr. Naifeng Hu, and the corresponding author was Prof. Guangxin Liang.
Beginning with a readily available
R
-carvone derivative, they developed a 3-step preparation of the tricyclic [5.5.6] caged structure featuring a crucial Pauson-Khand reaction, in the process setting an all-carbon quaternary center. A following Cu-catalyzed conjugate addition then stereoselectively installed the second all-carbon quaternary center. They then utilized a key retro-oxa-Michael/1, 2-addition/oxa-Michael cascade to introduce the olefin side chain. The third all-carbon quaternary center was then set at a late stage by alkylation of a 1, 3-dicarbonyl moiety. The synthesis of (-)-indoxamycin A was eventually accomplished in 13 steps. For the synthesis of (-)-indoxamycin B, they applied a Mukaiyama aldol reaction to introduce the requisite hydroxymethyl group, and the synthesis of this molecule took a total of 16 steps. It is noteworthy that the syntheses of enantiomeric (+)-indoxamycins A and B could be achieved by using
S
-carvone as the starting material. This provides the possibility of studying the biological functions of both enantiomers of indoxamycins. Modern medicinal chemistry has drawn ever increasing attention to the fact that enantiomeric products may lead to different biological activities. The ability to prepare both enantiomers of a target molecule is becoming progressively important.

Authors:Naifeng Hu, Changming Dong, Cuifang Zhang, Guangxin Liang
Orginal Article(scan or press the following QR code,this will lead to the original article after code recognition):

Total synthesis of (−)-Indoxamycins A and B
Angew. Chem. Int. Ed.,
2019, DOI: 10.1002/anie.201902043
PI Introduction
Prof. Guangxin Liang


